Contact
email: anstined@msu.edu
Dylan M. Anstine, Ph.D.
Dept. of Chem. Engineering and Mat. Science
Michigan State University
428 S. Shaw Lane, Room 1248
East Lansing, Michigan 48824-1226
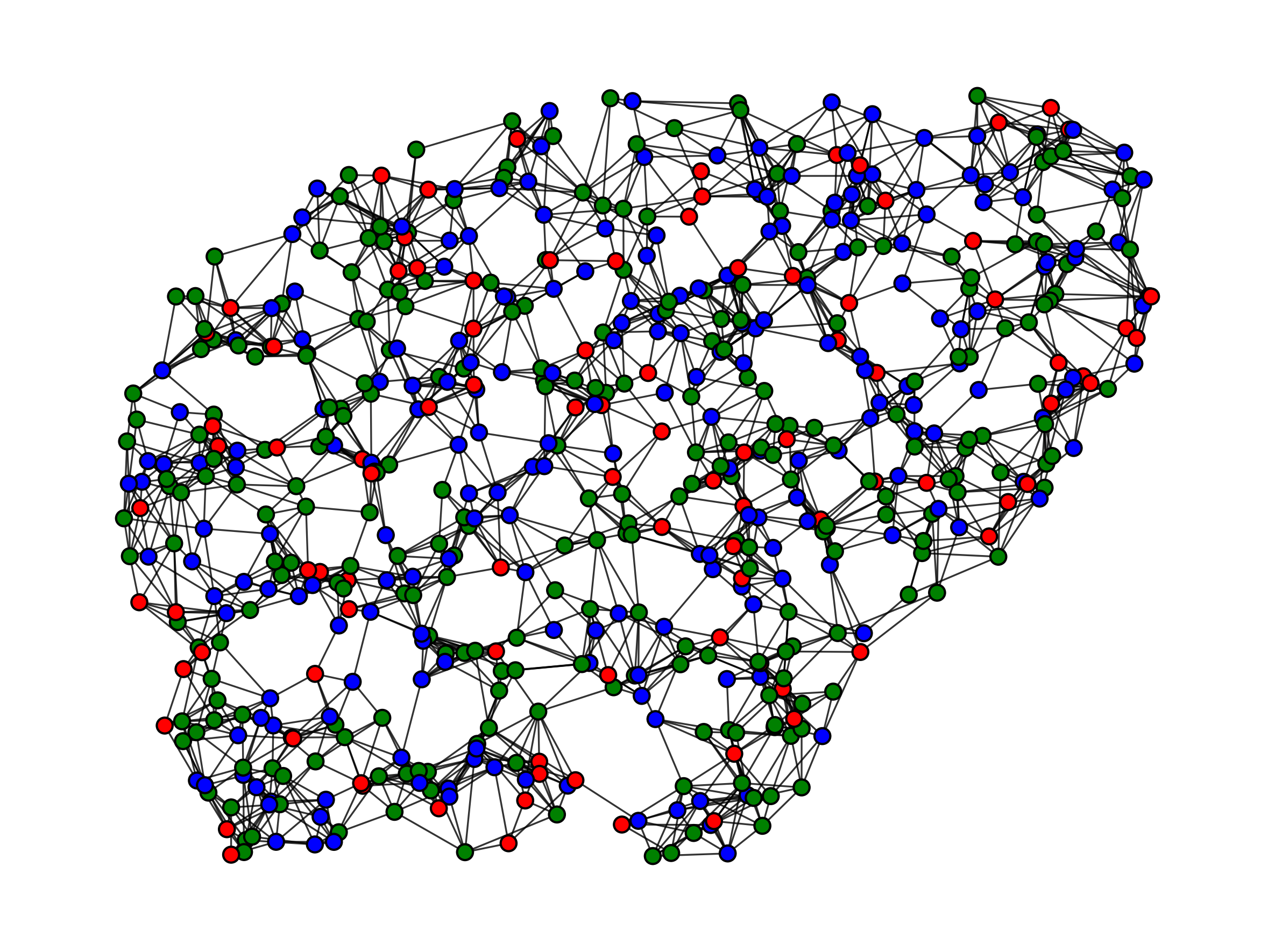
Frontier challenges in reaction planning, e.g., natural product synthesis, and materials with a facile end-of-life, particularly the degradation of polymers, are hindered by the enormity of possible reactions. The interconnectedness of multi-step pathways defining these areas give rise to deep reaction networks (DRNs), which routinely encompass a scale of reaction complexity that is beyond what can feasibly be analyzed by a human expert chemist. By framing reaction optimization as an action-reward system, The Anstine Research Lab develops innovative reinforcement learning approaches to transform sustainable polymers and human health by AI-exploited DRNs.

A ubiquitous challenge in developing functional polymers for ion exchange membranes, carbon capture, or chemical separations is to establish an accurate framework for monomer-to-polymer and polymer-to-property predictions. The microporous and amorphous nature of such membranes leads to difficulties in modeling their structure, validating predictions with experimental measurements, and simulating adsorption or transport. To overcome these limitations, the Anstine Lab develops tools for constructing realistic membrane models and identifies robust data-driven predictors that relate chemical identity to membrane performance. Our primary targets are exceptional classes of intrinsically microporous polymers, where the interconnected membrane void space arises from bulky and structurally rigid building blocks.
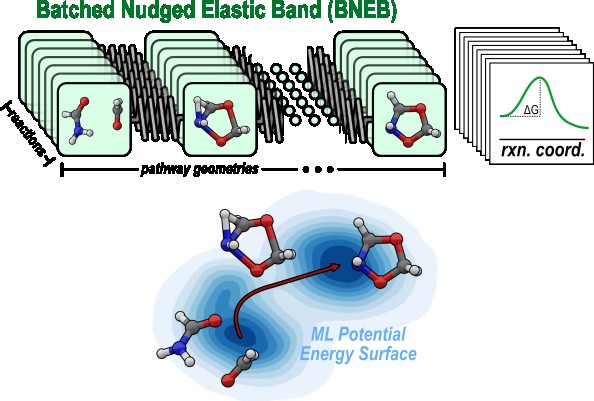
Clean energy and decarbonization efforts hinge critically on both heterogenous and homogenous catalyst compounds. Whether it be methanol synthesis, hydrogen production, or valorization of petroleum products, realizing such solutions on an industrial scale requires catalysts that achieve thermodynamic and kinetic performance targets while ideally operating under green conditions. Many attractive classes of catalysts exhibit an enormous number of possible compounds, leaving chemical engineers and materials scientists challenged to identify catalysts that meet these demands. The Anstine Lab champions a computational approach to catalyst design, performing high-throughput mechanistic modeling and deriving atomic-level design principles that would be otherwise infeasible to discover experimentally.
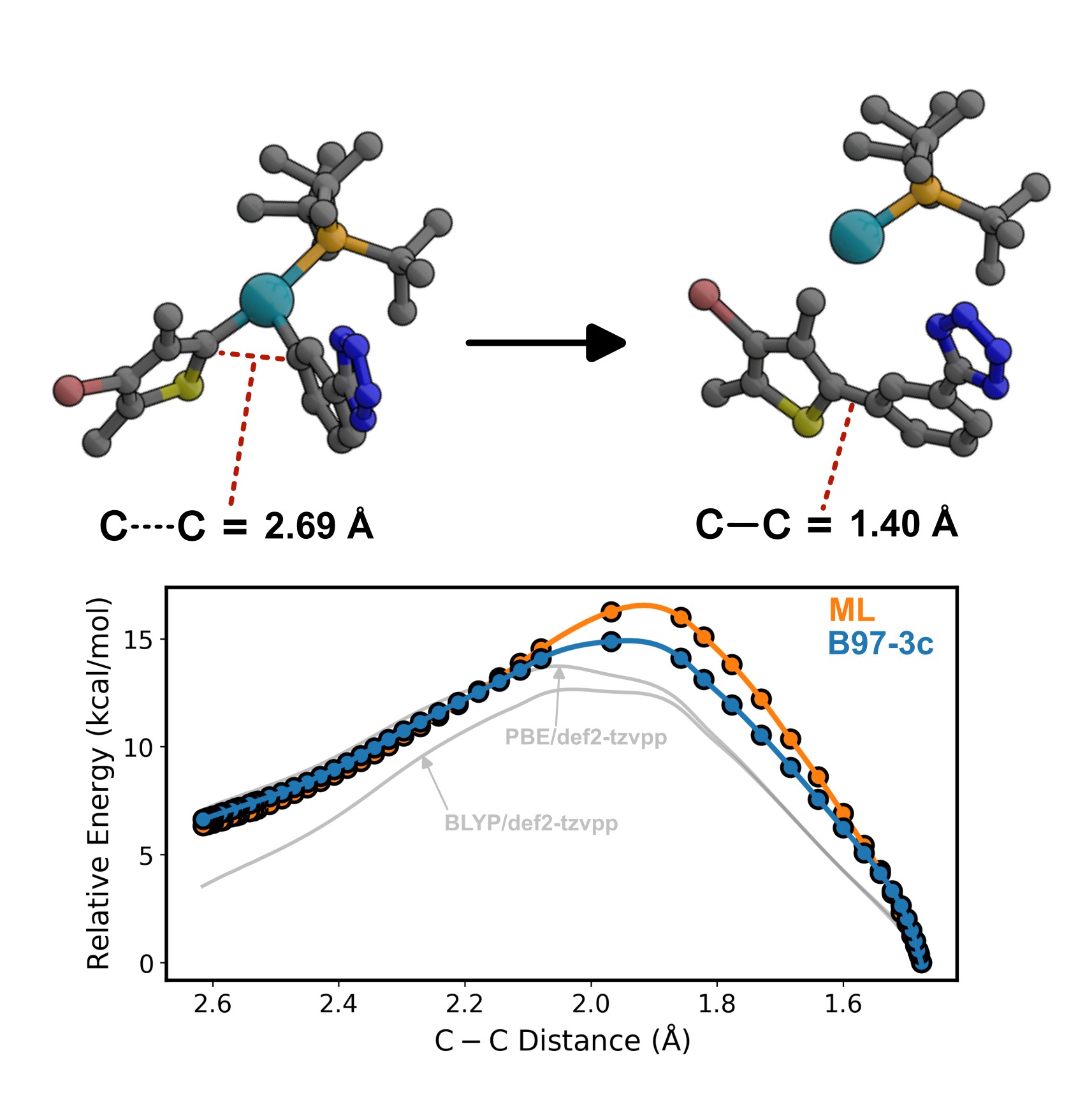
The tremendous advances in computing power, largely due to the growing sophistication of graphics processing units (GPUs), have created an opportunity to conduct molecular modeling on an unprecedented scale. Without exaggeration, it is now possible to perform thousands of molecular simulations, a workload of an entire Ph.D. one decade ago, over morning coffee. To extend this scale to billions of computational experiments per day, development of efficient algorithms and a tight integration of machine learning (ML) with chemistry is required. A core approach of the Anstine Lab to accomplish this is the development of machine learned interatomic potentials, allowing the accuracy of quantum chemistry to be accessed at scale with sub-second GPU times.
email: anstined@msu.edu
Dylan M. Anstine, Ph.D.
Dept. of Chem. Engineering and Mat. Science
Michigan State University
428 S. Shaw Lane, Room 1248
East Lansing, Michigan 48824-1226